Research Article 
![]() Creative Commons, CC-BY
Creative Commons, CC-BY
Quantum Chemical and Pharmacokinetic Studies of some Proton Pump Inhibitor Drugs
*Corresponding author: Moniruzzaman, Faculty of Engineering, Department of Applied Chemistry & Biochemical Engineering, Shizuoka University, 3-5-1, Johoku, Hamamatsu, 432-8561, Japan.
Received: February 28, 2019 Published: March 21, 2019
DOI: 10.34297/AJBSR.2019.02.000562
Abstract
Proton-pump inhibitor (PPIs) drugs are widely used in the treatment of gastric diseases. Almost all PPI drugs have some side effects depend on the physical condition and limit of doses. In this study, the physicochemical and pharmacokinetic studies of Omeprazole, Esomeprazole, Lansoprazole and Pantoprazole have been investigated. Density functional theory (DFT) with B3LYP/3-21g basis set has been employed to optimize the geometry and to elucidate their thermochemical, molecular orbital, molecular electrostatic potential properties. Pharmacokinetic parameters are also investigated to compare their absorption, distribution, excretion, metabolism, and toxicity.
Keywords: Proton-pump inhibitor; Density functional theory; Thermochemistry; HOMO-LUMO; Pharmacokinetic
Introduction
Omeprazole, lansoprazole, pantoprazole and esomeprazole having the benzimidazole nucleus and widely used in gastrointestinal treatment [1]. They play a significant role as proton pump inhibitors (PPIs) [2]. PPIs are not only very effective but also safer agents applied for the treatment of various gastrointestinal disorders, where they restrict the gastric acid production [3]. They deliver the best medication for gastroesophageal reflux disease (GERD) and erosive esophagitis through gastric acid suppressive therapy. Besides these drugs also used as a remedy of a helicobacter pylori infections. Generally, stomach acids are highly responsible for digesting nutrients whereas decreasing these acids can provide relief from heartburn and indigestion. Most importantly these drugs (Omeprazole, lansoprazole, pantoprazole and esomeprazole) are differ from each other in their metabolism and as well as in efficiency to interact with other prescripts. From more than a decade these drugs have been widely used as an acid inhibitory agent for treatment related to gastric acid secretion [4]. They irreversibly restrict the enzyme scheme by blocking the H+/K+ ATPase system [5]. Headaches, diarrhea, abdominal pain, vomiting etc. are the common side effects where long term use may rise the risk of gastric cancer and bone fracture as well [6-8]. These drugs have enriched availability in the markets all over the world. Omeprazole, the first developed drug in proton pump inhibitors category, is competitively maximum p-glycoprotein inhibitor rather than other PPIs [9,10]. It has already been reported for rapid medication of heartburn and acid reflux symptoms, Esomeprazole may be highly significant than other three agents [11].
In this study, we report the optimization of Omeprazole, Esomeprazole, Lansoprazole and Pantoprazole drugs to investigate their equilibrium geometry, thermal stability, dipole moment, chemical reactivity, potentiality and electrostatic potentiality. Pharmacokinetic predictions are also investigated to search their biochemical behavior i.e. absorption, metabolism and toxicity. A few drugs show developed thermal, molecular orbital and binding properties compared to others.
Methods and Materials
Computational details
Quantum mechanical methods are commonly used to calculate the thermochemical, molecular orbital and equilibrium geometry properties [12]. Initial geometry of all drugs was taken from online structure database named ChemSpider [13]. Density functional theory (DFT) along with B3LYP hybrid functional [14,15] under 3-21G basis set [16,17] have been employed to optimize all the drugs. Geometry optimization and another quantum calculation carried out using Gaussian09 program package [18].
Frontier molecular orbital features εHOMO and εLUMO were calculated at same level of theory. Chemical hardness (η), softness (S) and potential (μ) of all drugs were calculated from the energy of HOMOs and LUMOs considering Parr and Pearson interpretation [19,20] of DFT and Koopmans theorem [21] using following equations;
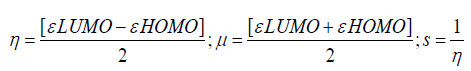
Pharmacokinetic studies
Admet SAR online database has been utilized to predict the absorption, distribution, metabolism, excretion and toxicity of all drugs [22]. Structure data file and simplified molecular-input line-entry system strings were used to calculate pharmacokinetic analysis.
Result and Discussion
Thermodynamic analysis
Molecular formula, molecular weight, enthalpy, free energy and dipole moment are depicted in (Table 1). Free energy is an important criterion to predict the reaction spontaneity [23]. Binding properties are influenced by free energy where, greater negative values are more favorable for spontaneous binding and interactions. Here it is found that the free energy of Omeprazole and Esomeprazole are same (-1439.246 Hartree), where Esomeprazole is the (S)- (-)-enantiomer of Omeprazole. The free energy of Lansoprazole -1621.511 Hartree where Pantoprazole shows the highest negative value -1672.379 Hartree. Increased negative value suggesting the better stability. Greater value of dipole moment improves the polar nature of a molecule [24]. The dipole moment of Omeprazole is 3.4410 Debye where Pantoprazole shows the highest dipole moment (9.6166 Debye).

Table 1: Molecular formula, molecular weight, enthalpy, free energy (in Hartree), and dipole moment (Debye) of all drugs.
Molecular orbital analysis
The HOMO and LUMO energies, gap, hardness, softness and chemical potential of all drugs are tabulated in (Table 2). Large HOMO-LUMO gap influence the high kinetic stability and low chemical reactivity, where small HOMO-LUMO gap is responsible for low chemical stability, because in addition of electrons to a highlying LUMO and/or removal of electrons from a low-lying HOMO is energetically insist the potential reaction [25]. In current study, large HOMO-LUMO gaps are found in Lansoprazole (5.064 eV) and Pantoprazole (5.048 eV). Where, Omeprazole and Esomeprazole have the lower energy gap (4.811eV) which suggesting the better chemical reactivity.

Table 2: HOMO, LUMO, gap, hardness, and softness of all drugs.
Molecular electrostatic potential analysis
Possible electrophilic and nucleophilic attack of chemical species can be predicted by molecular electrostatic potential (MEP) calculation. Biological recognition process as well as hydrogen bonding interactions between drugs and respected protein can be predicted by MEP calculation [26], where the red color denote maximum negative area and favorable site for electrophilic attack, blue color represent the maximum positive area which is favorable site for nucleophilic attack and green color represent for zero potential area. From MEP map, the maximum negative potentiality is found in Omeprazole (−0.2334 a.u.) and highest positive potentiality is observed in Pantoprazole (+0.4030 a.u.) (Figure 1 & 2).

Figure 1: Chemical and most stable optimized structure of all drugs.

Figure 2: Molecular electrostatic potential (a.u.) maps of all drugs calculated at 3-21G level theory.
Equilibrium geometry analysis
Selected bond distances and angles are summarized in (Table 3a & 3b) respectively (for atoms number see the optimized structures). In this study, drugs are optimized in gas phase. Calculated bond distances and angles were compared with experimental data to support the optimized structures [1,27]. From equilibrium geometry data it has been observed that the calculated bond distances and angles are approximately same with the experimental data which support the optimized structures.

Table 3: Theoretical and experimental (X-ray diffraction) (a) bond distances and (b) angles of all drugs.
Pharmacokinetic analysis
From pharmacokinetic data (Table 4), all the drugs show positive result to blood brain barrier (BBB) and human intestinal absorption which support absorption and distribution nature of all drugs. All the drugs are non-carcinogenic and exhibit group III acute oral toxicity which supporting the harmless properties for oral administration. All the drugs are P-glycoprotein noninhibitor where inhibition can interrupt the absorption, permeability and retention of drugs [28]. In addition, all the drugs are weak inhibitor to human ether-a-go-go-related gene (hERG) which can lead to long QT syndrome [29].

Table 4: Selected pharmacokinetic parameters of all drugs.
Conclusions
From the above quantum chemical calculations, Pantoprazole is thermally and configurationally more stable compare to other drugs with maximum dipole moment which suggesting better binding affinity and interactions with respected protein. From molecular orbital analysis, Omeprazole and Esomeprazole having low HOMO-LUMO gap with higher softness. Equilibrium geometry calculations also support the quantum calculations. Omeprazole and Pantoprazole are better for electrophilic and nucleophilic attack than two others. Pharmacokinetic study also forecast that all drugs are harmless and non-carcinogenic as well as safe for use. This study may be helpful to understand and compare the physicochemical and pharmacokinetic properties of these drugs.
Acknowledgement
Author would like to thank to Jannatun Nayeem, IIUC, Chittagong, Bangladesh for her support during data collection.
References
- Swamy G, Ravikumar K (2007) Crystal structure of lansoprazole sulfone. Journal of Structural Chemistry 48: 715-718.
- Siller Matula JM, Spiel AO, Lang IM, Gerhard Kreiner, Guenter Christ, et al. (2009) Effects of pantoprazole and esomeprazole on platelet inhibition by clopidogrel. American heart journal 157(1): 148.e1-148.e5.
- Welage LS, Berardi RR (2000) Evaluation of omeprazole, lansoprazole, pantoprazole, and rabeprazole in the treatment of acid-related diseases. Journal of the American Pharmaceutical Association 40(1): 52-62.
- Li X Q, Andersson TB, Ahlström M, Weidolf L (2004) Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug metabolism and disposition 32(80): 821-827.
- Sakai H, Fujii T, Takeguchi N (2016) Proton-potassium (H+/K+) ATPases: properties and roles in health and diseases. In: The Alkali Metal Ions: Their Role for Life 16: 459-483.
- Rossi S (2006) Australian medicines handbook. Adelaide: Australian Medicines Handbook p. 2-3.
- Cheung KS, Chan EW, Wong AYS (2018) Long-term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori: a population-based study 67: 28-35.
- Administration USF and D (2011) FDA drug safety communication: Possible increased risk of fractures of the hip, wrist, and spine with the use of proton pump inhibitors. Post market Drug Safety Information for Patients and Providers.
- Pauli Magnus C, Rekersbrink S, Klotz U, Fromm MF (2001) Interaction of omeprazole, lansoprazole and pantoprazole with P-glycoprotein. Naunyn-Schmiedeberg’s archives of pharmacology 364: 551-557.
- Lambrecht N, Corbett Z, Bayle D, Steve J D Karlish, George Sachs (1998) Identification of the site of inhibition by omeprazole of a α-β fusion protein of the H, K-ATPase using site-directed mutagenesis. Journal of Biological Chemistry 273: 13719-13728.
- Zheng RN (2009) Comparative study of omeprazole, lansoprazole, pantoprazole and esomeprazole for symptom relief in patients with reflux esophagitis. World Journal of Gastroenterology: WJG 15(8): 990- 995.
- Gleeson MP, Gleeson D (2009) QM/MM Calculations in Drug Discovery: A Useful Method for Studying Binding Phenomena? Journal of Chemical Information and Modeling 49(3): 670-677.
- Pence HE, Williams A (2010) ChemSpider: An Online Chemical Information Resource. Journal of Chemical Education 87(11): 1123- 1124.
- Becke AD, J Chem (1993) Phys 98: 5648.
- Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B 37(2): 785-789.
- Görling A, Trickey SB, Gisdakis P, Rösch N (1999) A critical assessment of density functional theory with regard to applications in organometallic chemistry. In: Organometallic Bonding and Reactivity. Springer pp. 109- 163.
- Parr RG, Szentpály L v, Liu S (1999) Electrophilicity index. Journal of the American Chemical Society 121(9): 1922-1924.
- Frisch MJ (2009) Guassian 09, Gaussian, Wallingford, CT.
- Calais JL (1993) Density-functional theory of atoms and molecules. R.G. Parr and W. Yang, Oxford University Press, New York, Oxford, 1989. IX + 333 pp International Journal of Quantum Chemistry 47: 101.
- Pearson RG (1995) The HSAB Principle more quantitative aspects. Inorganica Chimica Acta 240(1-2): 93-98.
- Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proceedings of the National Academy of Sciences 83(22): 8440-8441.
- Seeliger D, De Groot BL (2010) Conformational transitions upon ligand binding: holo-structure prediction from apo conformations. PLoS computational biology 6(1): e1000634.
- Cohen N, Benson SW (1993) Estimation of heats of formation of organic compounds by additivity methods. Chemical Reviews 93(7): 2419-2438.
- Lien EJ, Guo Z R, Li R L, Su C T (1982) Use of dipole moment as a parameter in drug-receptor interaction and quantitative structureactivity relationship studies. Journal of Pharmaceutical Sciences 71(6): 641-655.
- Aihara J (1999) Reduced HOMO−LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. The Journal of Physical Chemistry A 103(37): 7487-7495.
- Politzer P, Murray JS (1991) Molecular electrostatic potentials and chemical reactivity. Reviews in computational chemistry 273-312.
- Ohishi H, In Y, Ishida T, et al. (1989) Structure of 5-methoxy-2- {[4-methoxy-3, 5-dimethyl-2-pyridinylmethyl] sulfinyl}-1Hbenzimidazole (omeprazole). Acta Crystallographica Section C: Crystal Structure Communications 45: 1921-1923.
- Uzzaman M, Hoque MJ (2018) Physiochemical, molecular docking, and pharmacokinetic studies of Naproxen and its modified derivatives based on DFT. International Journal of Scientific Research and Management.
- Sanguinetti MC, Tristani-Firouzi M (2006) HERG potassium channels and cardiac arrhythmia. Nature 440: 463-469.


 We use cookies to ensure you get the best experience on our website.
We use cookies to ensure you get the best experience on our website.