Mini Review 
![]() Creative Commons, CC-BY
Creative Commons, CC-BY
Innovative Thinking for Rare Diseases Drug Development
*Corresponding author: Shein-Chung Chow, Department of Biostatistics and Bioinformatics, Duke University School of Medicine, 2424 Erwin Road, Hock Room 11037, Durham, NC 27705, USA.
Received: February 13,2020; Published: February 18, 2020
DOI: 10.34297/AJBSR.2020.07.001159
Abstract
For rare disease drug development, one of the major challenges is that there are only limited subject available for clinical trials. FDA (2019), however, indicated that the Agency does not have intention to create a statutory standard for rare diseases drug development. In this case, some out-of-the-box innovative thinking are necessary for obtaining substantial evidence for approval of rare disease drug product. The out-of-the-box innovative thinking include (i) probability monitoring procedure for sample size requirement, (ii) the concept of demonstrating not-ineffectiveness rather than demonstrating effectiveness, (iii) borrowing real-world data (RWD) in support of regulatory approval of rare diseases drug products, and (iv) the use of complex innovative design to shorten the process of drug development. Along this line, Chow and Huang (2020) proposed an innovative approach for rare diseases drug development by first demonstrating not-ineffectiveness with limited subjects available and then utilizing (borrowing) real-world data to rule out the probability of inconclusiveness for demonstration of effectiveness under a two-stage adaptive seamless trial design. The proposed innovative approach cannot only overcome the problem of small patient population for rare diseases, but also achieve the same standard for evaluation of drug products with common conditions.
Keywords: Probability Monitoring Procedure; Demonstrating Not-ineffectiveness; Real-World Data; Real-World Evidence; Two-Stage Adaptive Seamless Trial Design
Introduction
In the United States (US), a rare disease is defined as a condition that affects fewer than 200,000 people [1]. One of the most challenges for rare diseases drug development is probably the availability of subjects with the diseases under a small patient population. It is then of a great concern how to conduct clinical trials with the limited number of subjects available for obtaining substantial evidence regarding effectiveness and safety in support of regulatory approval of the rare disease drug product under investigation. For regulatory review and approval of rare disease (orphan) drug product, the US Food and Drug Administration (FDA) indicated that the Agency does not have intention to create a statutory standard for approval of orphan drugs that is different from the standard for approval of drugs in common conditions [2,3]. In this case, some out-of-the-box innovative thinking and approach for obtaining substantial evidence for approval of rare diseases drug products are necessarily applied.
In what follows, some out-of-the-box innovative thinking for rare disease drug (clinical) development are described. These out-ofthe- box innovative thinking include, but are not limited to,
(i) probability monitoring procedure for sample size calculation/ justification [4],
(ii) demonstrating not-ineffectiveness rather than demonstrating effectiveness based on limited number of subjects available of the small patient population of the rare disease under study [5],
(iii) utilizing (borrowing) real-world data (RWD), which generates real-world evidence (RWE) in support of regulatory approval of the rare disease drug development, and
(iv) the use of complex innovative design such as n-of-1 trial design, adaptive trial design, master protocols, and Bayesian approach.
In this article, in addition, an innovative approach for rare disease drug development that utilizing a two-stage adaptive seamless trial design in conjunction with the use of RWD/RWE is proposed [5]. The proposed innovative approach cannot only overcome the problem of small patient population for rare diseases, but also achieve the same standard for evaluation of drug products with common conditions.
Innovative Thinking for Rare Disease Drug Development
To overcome the problem of small patient population for rare diseases, some out-of-the-box innovative thinking regarding design (sample size requirement and complex innovative trial design) and analysis (e.g., demonstration of effectiveness and the utilization of RWD/RWE) are necessarily considered.
Probability Monitoring Procedure for Sample Size
For rare disease clinical development, it is recognized that a pre-study power analysis for sample size calculation is not feasible due to the fact that there are only limited number of subjects available for the intended trial, especially when the anticipated treatment effect is relatively small and/or the variability is relatively large. In this case, alternative methods such as precision analysis (or confidence interval approach), reproducibility analysis, and probability monitoring approach may be considered for providing substantial evidence with certain statistical assurance [6]. It, however, should be noted that the resultant sample sizes from these different analyses could be very different with different levels of statistical assurance achieved. Thus, for rare disease clinical trials, it is suggested that an appropriate sample size should be selected for achieving certain statistical assurance under a valid trial design. To overcome the problem, [4] proposed a probability monitoring procedure for sample size calculation/justification, which can substantially reduce the required sample size for achieving certain statistical assurance.
As an example, an appropriate sample size may be selected
based on a probability monitoring approach such that the probability
of crossing safety boundary is controlled at a pre-specified
level of significance. Suppose an investigator plans to monitor the
safety of a rare disease clinical trial sequentially at several times,AS 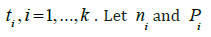 be the sample size and the probability
of observing an event at time
ti . Thus, an appropriate sample
size can be selected such that the following probability of crossing
safety stopping boundary is less than a pre-specified level of significance.
be the sample size and the probability
of observing an event at time
ti . Thus, an appropriate sample
size can be selected such that the following probability of crossing
safety stopping boundary is less than a pre-specified level of significance.

Note that the concept of the probability monitoring approach should not be mixed up the concepts with those based on power analysis, precision analysis, and reproducibility analysis. That is, Statistical methods for data analysis should reflect the desired statistical assurance under the trial design.
Demonstrating Not-Ineffectiveness Versus Demonstrating Effectiveness
For approval of a new drug product, the sponsor is required to provide substantial evidence regarding safety and efficacy of the drug product under investigation. In practice, a typical approach is to conduct adequate and well-controlled clinical studies and test the following point hypotheses:
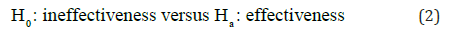
The rejection of the null hypothesis of ineffectiveness is in favor of the alternative hypothesis of effectiveness. Most researchers interpret the rejection of the null hypothesis is the demonstration of the alternative hypothesis of effectiveness. It, however, should be noted that “in favor of effectiveness” does not imply “the demonstration of effectiveness”. In practice, hypothesess (2) should be
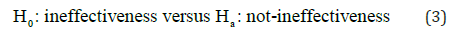

Figure 1: Relationship between “not ineffectiveness” and “effectiveness”.
In other words, the rejection of H0 would lead to the conclusion of “not H0”, which is Ha i.e.,“not-ineffectiveness” as given in (3). As can be seen from Hain (2) and (3), the concept of effectiveness (2) and the concept of not ineffectiveness (3) are not the same. Not ineffectiveness does not imply effectiveness in general. Thus, the traditional approach for clinical evaluation of the drug product under investigation can only demonstrate “not-ineffectiveness” but not “effectiveness”. The relationship between demonstrating “effectiveness” (2) and demonstrating “not-ineffectiveness” (3) is illustrated in Figure 1. As it can be seen from (Figure 1), “not-ineffectiveness” consists of two parts, namely, the portion of “inconclusiveness” and the portion of “effectiveness”. As a result, the rejection of the null hypothesis of ineffectiveness cannot directly imply that the drug product is effective unless the probability of inconclusiveness, denoted by IC pic, is negligible, i.e.,
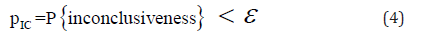
where ε is a pre-specific number which is agreed upon between clinician and regulatory reviewer [7].
Note that the concept of demonstrating “not ineffectiveness” is similar to that of establishing non-inferiority. One can test for superiority (i.e., effectiveness) once the non-inferiority has been established without paying any statistical penalties.
The Use of RWD/RWE
The 21st Century Cures Act passed by the United States Congress in December 2016 requires that the FDA shall establish a program to evaluate the potential use of real-world evidence (RWE) which is derived from real-world data (RWD) to (i) support approval of new indication for a drug approved under section 505 (c) and (ii) satisfy post-approval study requirements. RWD refers to data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources. RWD sources include, but are not limited to, electronic health record (EHR), administrative claims and enrollment, personal digital health applications, public health databases, and emerging sources. Although RWE offers the opportunities to develop robust evidence using high quality data and sophisticated methods for producing causal effect estimates regardless randomization is feasible. In this section, we have demonstrated that the assessment of treatment effect (RWE) based on RWD could be biased due to potential selection and information biases of RWD. Although fit-for-purpose RWE may meet regulatory standards under certain assumptions, it is not the same as substantial evidence (current regulatory standard). In practice, it is then suggested that when there are gaps between fit-for-purpose RWE and substantial evidence, we should make efforts to fill the gaps for an accurate and reliable assessment of the treatment effect.
In order to map RWE to substantial evidence (current regulatory standard), we need to have good understanding of the RWD in terms of data relevancy/quality and its relationship with substantial evidence so that a fit-for-regulatory purpose RWE can be derived to map to regulatory standard.
As indicated by Corrigan Curay [8] there is a value of using RWE to support regulatory decisions in drug review and approval process. However, incorporating RWE into evidence generation, many factors must be considered at the same time before we can map RWE to substantial evidence (current regulatory standard) for regulatory review and approval. These factors include, but are not limited to, (i) efficacy or safety, (ii) relationship to available evidence, (iii) clinical context, e.g., rare, severe, life-threatening, or unmet medical need, and (iv) natural of endpoint/concerns about bias. In addition, leveraging RWE to support new indications and label revisions can help accelerate high quality RWE earlier in the product lifecycle, providing more relevant evidence to support higher quality and higher value care for patients. Incorporating RWE into product labeling can lead to better-informed patient and provider decisions with more relevant information. For this purpose, it is suggested characterizing RWD quality and relevancy for regulatory purposes. Ultimate regulatory acceptability, however, will depend upon how robust these studies can be. That is, how well they minimize the potential for bias and confounding.
Complex Innovative Design
As indicated earlier, small patient population is a challenge to rare disease clinical trials. Thus, there is a need for innovative trial designs in order to obtain substantial evidence with small number of subjects available for achieving the same standard for regulatory approval. In this sub-section, several innovative trial designs including n-of-1 trial design, an adaptive trial design, master protocols, and a Bayesian design are discussed.
One of the major dilemmas for rare diseases clinical trials is the in-availability of patients with the rare diseases under study. In addition, it is unethical to consider a placebo control in the intended clinical trial. Thus, it is suggested an n-of-1 crossover design be considered. An n-of-1 trial design is to apply n treatments (including placebo) in an individual at different dosing periods with sufficient washout in between doing periods. A complete n-of-1 trial design is a crossover design consists of all possible combinations of treatment assignment at different dosing periods.
Another useful innovative trial design for rare disease clinical trials is an adaptive trial design. In its draft guidance on adaptive clinical trial design, FDA defines an adaptive design as a study that includes a prospectively planned opportunity for modification of one or more specified aspects of the study design and hypotheses based on analysis of (usually interim) data from subjects in the study [9, 10]. The FDA guidance has been served as an official document describing the potential use of adaptive designs in clinical trials since it was published in 2019. It, however, should be noted that the FDA draft guidance on adaptive clinical trial design is currently being revised in order to reflect pharmaceutical practice and FDA’s current thinking. [11] introduced the concept of master protocol for studying multiple therapies, multiple diseases, or both in order to answer more questions in a more efficient and timely fashion. Master protocols include the following types of trials: umbrella, basket, and platform. The type of umbrella trial is to study multiple targeted therapies in the context of a single disease, while the type of basket trial is to study a single therapy in the context of multiple diseases or disease subtypes. The platform is to study multiple targeted therapies in the context of a single disease in a perpetual manner, with therapies allowed to enter or leave the platform on the basis of decision algorithm. As indicated by [11], if designed correctly, master protocols offer a number of benefits include streamlined logistics, improved data quality, collection and sharing, as well as the potential to use innovative statistical approaches to study design and analysis. Master protocols may be a collection of sub-studies or a complex statistical design or platform for rapid learning and decision-making.
Under the assumption that historical data (e.g., previous studies or experience) are available, Bayesian methods for borrowing information from different data sources may be useful. These data sources could include, but are not limited to, natural history studies and expert’s opinion regarding prior distribution about the relationship between endpoints and clinical outcomes. The impact of borrowing on results can be assessed through the conduct of sensitivity analysis. One of the key questions of particular interest to the investigator and regulatory reviewer is that how much to borrow in order to (i) achieve desired statistical assurance for substantial evidence, and (ii) maintain the quality, validity, and integrity of the study.
Innovative Approach
Combining the out-of-the-box innovative thinking regarding rare disease drug development described in the previous section, [7] proposed the following innovative approach utilizing a twostage adaptive approach in conjunction with the use of RWD/RWE for rare diseases drug development. This innovative approach is briefly summarized below.
Step 1: Select a small sample size n1 at Stage 1 as deemed appropriate by the principal investigator (PI) based on both medical and non-medical considerations. Note that n1 may be selected based on the probability monitoring procedure.
Step 2: Test hypotheses (3) for not-ineffectiveness at the α1 level, a pre-specified level of significance. If fails to reject the null hypothesis of ineffectiveness, then stop the trial due to futility. Otherwise proceed to the next stage. Note that an appropriate value of α1 can be determined based on the evaluation of the trade-off with the selection of α 2for controlling the overall type I error rate at the significance level of α . The goal of this step is to establish non-inferiority (i.e., not-ineffectiveness) of the test treatment with limited number of subjects available at the α1 level of significance based on the concept of probability monitoring procedure for sample size justification and performing a non-inferiority (not-ineffectiveness) test with a significance level of α1
Step 3a: Recruit additional n2 subjects at Stage 2. Note that n2 may be selected based on the probability monitoring procedure. Once the non-inferiority (not ineffectiveness) has been established at Stage 1, sample size re-estimation may be performed for achieving the desirable statistical assurance (say 80% power) for establishment of effectiveness of the test treatment under investigation at the second stage (say n2 , sample size required at Stage 2).
Step 3b: Obtain (borrow) n2 − n2 data from previous studies (real-world data) if the sample size of n2 subjects are not enough for achieving desirable statistical assurance (say 80% power) at Stage 2. Note that data obtained from the n2subjects are from randomized clinical trial (RCT), while data obtained from the other n2 − n2 are from RWD.
Step 4: Combined data from both Step 3a (data obtained from RCT) and Step 3b (data obtained from RWD) at Stage 2, perform a statistical test to eliminate the probability of inconclusiveness. That is, perform a statistical test to determine whether the probability of inconclusiveness has become negligible at the α2 level of significance. For example, if the probability of inconclusiveness is less than a pre-specified value (say 5%), we can then conclude the test treatment is effective.
In summary, for review and approval of rare diseases drug products, Chow and Huang (2020) proposed first to demonstrate not-ineffectiveness with limited information (patients) available at a pre-specified level of significance of α1. Then, after the not-ineffectiveness of the test treatment has been established, collect additional information (real-world data) to rule out the probability of inconclusiveness for demonstration of effectiveness at a pre-specified level of significance of α2 under the two-stage adaptive seamless trial design.
Concluding Remarks
In this article, some out-of-the-box innovative thinking regarding rare disease drug development are described. These innovative thinking include (i) probability monitoring procedure for sample size calculation/justification for certain statistical assurance, (ii) the concept of testing non-inferiority (i.e., demonstrating notineffectiveness) with limited number of subjects available, (iii) utilizing (borrowing) real-world data (RWD) from various of data sources in support of regulatory approval of rare diseases drug products, and (iv) the use of a two-stage adaptive seamless trial design to shorten the process of drug development. Combining these innovative thinking [12, 13], under a two-stage adaptive seamlesstrial design, [7] proposed an innovative approach for rare diseases drug development by first demonstrating not-ineffectiveness based on limited subjects available and then utilizing (borrowing) real-world data to rule out the probability of inconclusiveness for demonstration of effectiveness. Chow and Huang’s proposed innovative approach cannot only overcome the problem of small patient population for rare diseases, but also achieve the same standard for evaluation of drug products with common conditions.
References
- ODA (1983) Orphan Drug Act of 1983. Pub L No 97-414.
- FDA (2015) Guidance for Industry - Rare Diseases: Common Issues in Drug Development. Center for Drug Evaluation and Research, the U.S. Food and Drug Administration, Silver Spring, Maryland, USA.
- FDA (2019a) Guidance for Industry – Rare Diseases: Common Issues in Drug Development. The U.S. Food and Drug Administration, Silver Spring, Maryland, USA.
- Huang Z, Chow SC (2019) Probability monitoring procedure for sample size determination. Journal of Biopharmaceutical Statistics 29: 887-896.
- Chow SC, Huang Z (2019) Demonstrating effectiveness or demonstrating not ineffectiveness - A potential solution for rare disease drug development. Journal of Biopharmaceutical Statistics 29(5): 897-907.
- Chow SC, Shao J, Wang H, Lokhnygina (2017) Sample Size Calculations in Clinical Research. 3rd Edition, Chapman and Hall/CRC Press, Taylor & Francis, New York, USA.
- Chow SC, Huang Z (2020) Innovative approach for rare disease drug development. Journal of Biopharmaceutical Statistics 30(3).
- Corrigan Curay J (2018) Real-world evidence – FDA update. Presented at RWE Collaborative Advisory Group Meeting, Duke-Margolis Center, Washington DC, USA.
- FDA (2010) Draft Guidance for Industry – Adaptive Design Clinical Trials for Drugs and Biologics. United States Food and Drug Administration, Silver Spring, Maryland, USA.
- FDA (2019c) Adaptive Designs for Clinical Trials of Drugs and Biologics. United States Food and Drug Administration, Silver Spring, Maryland, USA.
- Woodcock J, LaVange LM (2017) Master protocols to study multiple therapies, multiple diseases, or both. N Eng J Med 377(1): 62-70.
- FDA (2018) Guidance for Industry – Master Protocols: Efficient Clinical Trial Design Strategies to Expedite Development of Oncology Drugs and Biologics. The United States Food and Drug Administration, Silver Spring, Maryland, USA.
- FDA (2019b) Framework for FDA’s Real-World Evidence Program. United States Food and Drug Administration, Silver Spring, Maryland, USA.


 We use cookies to ensure you get the best experience on our website.
We use cookies to ensure you get the best experience on our website.