Research Article 
![]() Creative Commons, CC-BY
Creative Commons, CC-BY
New Nano-Molecule Kurumi and Raman Spectroscopy using ab initio, Hartree-Fock Method
*Corresponding author: Ricardo Gobato, Laboratory of Biophysics and Molecular Modeling Genesis, State Secretariat for Education of Parana, Bela Vista do Paraiso, Parana, Brazil.
Received: January 25, 2019; Published: April 18, 2019
DOI: 10.34297/AJBSR.2019.02.000594
Abstract
The work characterizes the Raman spectrum of the new nano-molecule C13H20BeLi2SeSi /C13H19BeLi2SeSi , nano-molecule Kurumi. Calculations obtained in the methods Restrict Hartree-Fock of the first principles (ab initio), on the set of basis used indicate that the simulated molecule C13H20BeLi2SeSi /C13H19BeLi2SeSi features the structure polar-apolar-polar predominant. The set of basis used that have are correlation-consistent polarized Triple-zeta (CC-pVTZ) and Pople’s basis sets six gaussian functions in the shell, three double zeta Gaussian functions, Slater type orbitals with polarization function (6-311G** (3df, 3pd)). In the CC-pVTZ base set, the charge density in relation to 6-311G** (3df, 3pd) is 50% lower. The length of the molecule C13H20BeLi2SeSi is of 15.799Å. The Raman spectrum was calculated indicating the characteristic of the nano-molecule and their frequency (cm-1) were obtained in the set of bases used. The highest for Raman scattering activities peaks are in the frequency 3,348cm-1 with 7.107609729 Å4/amu and 2,163 cm-1 with 8.902805583 Å4/amu, for CC-pVTZ and 6-311G** (3df, 3pd), respectively. As the bio-inorganic molecule C13H20BeLi2SeSi is the basis for a new creation of a biomembrane, later calculations that challenge the current concepts of biomembrane should advance to such a purpose. The new nano-molecule Kurumi is well characterize computationally. As its scientific name 3-lithio-3-(6- {3-selena-8- beryllatricyclo [3.2.1.0²,⁴] oct-6-en-2-yl} hexyl) -1-sila-2-lithacyclopropane.
Keywords: Biomembrane; CC-pVTZ ; Hartree-Fock method; Nano-molecule Kurumi; Raman spectroscopy; 6-311G** (3df, 3pd)
Introduction
The structure of the C13H20BeLi2SeSi is a bio-inorganic seed molecule for a biomembrane genesis that defies the current concepts of a protective mantle structure of a cell such as biomenbrane [1-3] to date is promising, challenging. Leaving to the Biochemists their experimental synthesis. Structures of a liquid crystal such as a new membrane may occur, micelles [1].
A large investigative study in the literature was carried out, but was not found of the structure studied here. There is an absence of a referential of the theme, finding only one work in [1]. The present study is an extension of the work already begun in the creation of ab initio, computational methods of quantum chemistry, to prove the possible structure of the bio-inorganic nano-molecule genesis, C13H20BeLi2SeSi . Their characterization by well-known ab initio methods of extreme precision is essential for their future experimental achievement. Therefore, the ab initio study using RHF (Restrict Hartree-Fock method) [4-13] and the set of basis used is CC-pVTZ [10-14] and 6-311G** (3df, 3pd) [14-37].
The calculation of the Raman spectrum of the nano-molecule Genesis has the purpose of deepening the knowledge and charac terization of this. The work characterizes the Raman spectrum of the nano-molecule C13H20BeLi2SeSi . Calculations obtained in the ab initio RHF (Restrict Hartree-Fock method) [4-13]. The set of basis used that have are CC-pVTZ [10-14] and 6-311G** (3df, 3pd) [7, 14-18].
Materials and Methods
Hartree-fock methods
The Hartree-Fock self–consistent method [4-13] is based on the one-electron approximation in which the motion of each electron in the effective field of all the other electrons is governed by a one-particle Schrodinger¨ equation. The Hartree-Fock approximation takes into account of the correlation arising due to the electrons of the same spin, however, the motion of the electrons of the opposite spin remains uncorrelated in this approximation. The methods beyond self-consistent field methods, which treat the phenomenon associated with the many-electron system properly, are known as the electron correlation methods. One of the approaches to electron correlation is the Møller-Plesset (MP) [4-18] perturbation theory in which the Hartree-Fock energy is improved by obtaining a perturbation expansion for the correlation energy. [4,5] However, MP calculations are not variational and can produce an energy value below the true energy [4,5]. The exchange-correlation energy is expressed, at least formally, as a functional of the resulting electron density distribution, and the electronic states are solved for self-consistently as in the Hartree-Fock approximation [4-13].
The full Hartree-Fock equations are given by
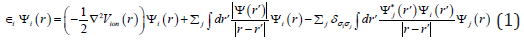
The minimal basis sets are not flexible enough for accurate representation of orbitals. Using multiple functions to represent each orbital, have the double-zeta basis set allows us to treat each orbital separately when we conduct the Hartree-Fock calculation:
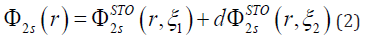
In Equation (2) 2s atomic orbital expressed as the sum of two STOs (Slater type orbitals) [4,5]. The two equations are the same except for the value of ξ which accounts for how large the orbital is. The constant d determines how much each STO will count towards the final orbital [4,5].
The vast literature associated with these methods suggests that the follwing is a plausible hierarchy :
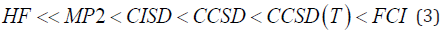
The extremes of ‘best’, FCI, and ‘worst’, HF, are irrefutable, but the intermediate methods are less clear and depend on the type of chemical problem being addressed. The use of HF [4-13] in the case of FCI was due to the computational cost.
Hardware and Software
For calculations a computer models was used: Intel CoreTM i3-3220 CPU @ 3.3 GHz x 4 processors [38], Memory DDR3 4GB, HD SATA WDC WD7500 AZEK-00RKKA0 750.1GB and DVD-RAM SATA GH44NS9 ATAPI, Graphics Intel Ivy Bridge [1].
The ab initio calculations have been performed to study the equilibrium configuration of C13H20BeLi2SeSi molecule using the GAMESS [14,15]. The set of programs Mercury 3.8 [39], Avogadro [40,41] are the advanced semantic chemical editor, visualization, and analysis platform and GAMESS [14,15] is a computational chemistry software program and stands for General Atomic and Molecular Electronic Structure System [14,15] set of programs.
For calculations of computational dynamics, the Ubuntu Linux version 16.10 system was used [42]. The Graphic was edited in Origin software [43], for comparison of the spectra obtained in the set of bases used.
Results
Our results takes place from the findings of the molecular dynamics of bio-inorganic nano-molecule C13H20BeLi2SeSi . The results were summarized and shown in Figures 1 & 2, Tables 1 & 2.
Nano-molecule Kurumi
The (Figure 1) representation of the molecular structure of C13H20BeLi2SeSi /C13H19BeLi2SeSi , Nano-molecule Kurumi, 3-lithio-3-(6-{3-selena-8-beryllatricyclo[3.2.1.0²,⁴]oct-6-en-2- yl}hexyl)-1-sila-2 lithacyclopropane.

Figure 1: Representation of the molecular structure of C13H19BeLi2SeSi , Nano-molecule Kurumi, 3-lithio-3-(6-{3-selena-8-beryllatricyclo[3.2.1.0²,⁴] oct-6-en-2-yl}hexyl)-1-sila-2-lithacyclopropane, obtained through computer via ab initio calculation method RHF/CC-pVTZ.
Raman spectrum
In (Figure 2) we have representation of the Raman spectrum of C13H20BeLi2SeSi - Nano-molecule Kurumi, 3-lithio-3-(6-{3-selena- 8-beryllatricyclo[3.2.1.0²,⁴]oct-6-en-2-yl}hexyl)-1-sila-2-lithacyclopropane.

Figure 2: Representation of the Raman spectrum of C13H20BeLi2SeSi - Frequency (cm-1) for Raman scattering activities (SR, Å4/amu) -using computer programs GAMESS, in the ab initio for the RHF method, in sets of basis RHF/CC-pVTZ in left and 6-311G** (3df, 3pd) in right.
Frequency (cm-1) for Raman scattering activities (SR, Å4/amu) -using computer programs GAMESS, in the ab initio for the RHF method, in sets of basis RHF/CC-pVTZ in left and 6-311G**(3df, 3pd) in right, obtained using computer software GAMESS. The Graphic edited in Origin software, for comparison of the spectra obtained in the set of bases used.
RHF/CC-pVTZ
The (Table 1) contains the frequencies (cm-1) for Raman scattering activities (SR, Å4/amu) of the Nano-molecule Kurumi, 3-lithio-3-(6-{3-selena-8-beryllatricyclo[3.2.1.0²,⁴]oct-6-en-2-yl} hexyl)-1-sila-2-lithacyclopropane.via ab initio methods, set base RHF/CC-pVTZ for the infrared spectrum.
The highest for Raman scattering activities peaks are in the frequency 708 cm-1 with 8.448529382 Å4/amu, 732 cm-1 with 6.968083358 Å4/amu, 1,180 cm-1 with 8.947657695 Å4/ amu, 2,180 cm-1 with 7.796950601 Å4/amu and 3,348 cm-1 with 7.107609729 Å4/amu for RHF/CC-pVTZ.
RHF/6-311G**(3df,3pd)
The (Table 2) contains the frequencies (cm-1) for Raman scattering activities (SR, Å4/amu) of the C13H20BeLi2SeSi molecule, of the Nano-molecule Kurumi, via ab initio methods, set base RHF/6- 311G**(3df,3pd) for the infrared spectrum.
The highest for Raman scattering activities peaks are in the frequency 602 cm-1 with 7.279393384 Å4/amu, 707 cm-1 with 7.719121469 Å4/amu, 2,079 cm-1 with 6.915053417 Å4/ amu, 2,163 cm-1 with 8.902805583 Å4/amu and 3,094 cm-1 with 6.213759835 Å4/amu for RHF/6-311G** (3df, 3pd).
Discussion
The calculations made so far admit a seed molecule at this stage of the quantum calculations of the arrangement of the elements we have chosen, obtaining a highly reactive molecule with the shape polar-apolar-polar. Its structure has polarity at its ends, having the characteristic polar-apolar-polar. Even using a simple base set the polar-apolar-polar characteristic is predominant. The set of bases used that have the best compatible, more precise results are CCpVTZ and 6-311G (3df, 3pd). In the CC-pVTZ base set, the charge density in relation to 6-311G (3df, 3pd) is 50% lower. The structure of the bio-inorganic seed molecule for a biomembrane genesis that challenge the current concepts of a protective mantle structure of a cell such as biomembrane to date is promising, challenging. Leaving to the biochemists their experimental synthesis.
The study evolves to construct a biomembrane from the seed molecule. The calculations default already performed admit a hydrophobic and hydrophilic molecule, the basis of the formation of a micelle and or biomembrane, as the default template. But kurumi molecule is polar-apolar-polar characteristic is predominant, i.e. A hydrophilic-hydrophobic-hydrophilic molecule, thus presenting another standard for the construction of a biomembrane, which is even more innovative because a bio-inorganic molecule. Going beyond imagination, the most innovative and challenging proposal of the work advances the construction of a structure compatible with the formation of a «new DNA», based now on the seed molecule. Our discussion takes place from the findings of the molecular dynamics of bio-inorganic nano-molecule C13H20BeLi2SeSi . The results were summarized and shown in Figures 1 & 2, Tables 1 & 2.
The Figure 1 representation of the molecular structure of C13H19BeLi2SeSi , Nano-molecule Kurumi, 3-lithio-3-(6-{3-selena- 8-beryllatricyclo[3.2.1.0²,⁴]oct-6-en-2-yl}hexyl)-1-sila-2-lithacyclopropane, obtained through computer via ab initio calculation method RHF/CC-pVTZ.
The Figure 2 represents of the Raman spectrum of C13H40Be- Li2SeSi - Frequency (cm-1) for Raman scattering activities (SR, Å4/amu) - using computer programs GAMESS, in the ab initio for the Restrict Hartree-Fock method, in sets of basis CC-pVTZ and 6-311G**(3df, 3pd), obtained using computer software GAMESS. Graphic edited in Origin software, for comparison of the spectra obtained in the set of bases used.
The Tables 1 & 2 present the Raman spectrum frequencies for CC-pVTZ and 6-311G** (3df, 3pd), respectively, for Raman scattering activities (SR, Å4/amu), SR > 1.
The highest for Raman scattering activities peaks are in the frequency 3,348 cm-1 with 7.107609729 Å4/amu and 2,163 cm-1 with 8.902805583 Å4/amu, for CC-pVTZ and 6-311G** (3df, 3pd), respectively.

Table 1: Table contains the frequencies (cm-1) for Raman scattering activities (SR, Å4/amu) of the C13H20BeLi2SeSi molecule via ab initio methods, set base RHF/CC-pVTZ for the infrared spectrum.

Table 2: Table contains the frequencies (cm-1) for Raman scattering activities (SR, Å4/amu) of the C13H20BeLi2SeSi molecule via ab initio methods, set base RHF/6-311G**(3df,3pd) for the infrared spectrum.
It presents “fingerprint” between the intermediate frequency intervals presented in Tables 1 & 2.
Calculations obtained in the ab initio RHF method, on the set of basis used, indicate that the simulated molecule, C13H20BeLi2SeSi , is acceptable by quantum chemistry. Its structure has polarity at its ends, having the characteristic polar-apolar-polar.
The 6-311G** (3df, 3pd) set of basis exhibits the characteristic of the central chain, with a Small density of negative charges, Near the ends of the Carbons of this. In the CC-pVTZ base set, the charge density in relation to 6-311G (3df, 3pd) is 50% lower. It is characterized infrared spectrum of the molecule C13H20BeLi2SeSi , for absorbance and transmittance, in Hartree-Fock method in the set of bases CC-pVTZ and 6-311G (3df, 3pd). The dipole moments CC-pTZV are 3.69% bigger than 6-311G** (3df, 3pd).
The new nano-molecule Kurumi is well characterize computationally. As its scientific name 3-lithio-3-(6-{3-selena-8-beryllatricyclo[ 3.2.1.0²,⁴]oct-6-en-2-yl}hexyl)-1-sila-2-lithacyclopropane. Getting Kurumi name, which means boy in Tupi-Guarani language, which are indigenous inhabitants of southern Brazil.
Limitations
Our study has so far been limited to computational simulation via quantum chemistry, an applied theory. Our results and calculations are compatible and with the theory of quantum chemistry, but their physical experimental verification depends on advanced techniques for their synthesis, obtaining laboratory for experimental biochemical.
Conclusion
The highest for Raman scattering activities peaks are in the frequency 3,348 cm-1 with 7.107609729 Å4/amu and 2,163 cm-1 with 8.902805583 Å4/amu, for CC-pVTZ and 6-311G** (3df, 3pd), respectively. The Raman spectrum was calculated, indicating the characteristic of the bioinioganic nano-molecule genesis. Now the challenge is to build the basic structure of the bio-inorganic membrane. From the unimaginable, going where our mind can take us and build a new DNA, that nanomollecule. Characterized its infrared spectrum and Raman. Quantically calculated, accepted by quantum chemistry parameters, with ab initio methods, in the bases CC-pVTZ and 6-311G ** (3df, 3pd). An experimental challenge to chemists. The new bio-inorganic nano-molecule C13H20BeLi2SeSi / C13H19BeLi2SeSi , is well characterize computationally. As its scientific name 3-lithio-3-(6-{3-selena-8-beryllatricyclo[3.2.1.0²,⁴]oct- 6-en-2-yl}hexyl)-1-sila-2-lithacyclopropane, getting Kurumi name, which means boy in Tupi-Guarani language, which are indigenous inhabitants of southern Brazil. As the bio-inorganic nano-molecule Kurumi is the basis for a new creation of a biomembrane, later calculations that challenge the current concepts of biomembrane should advance to such a purpose.
Acknowledgement
None.
Conflict of Interest
No conflict of interest.
References
- R Gobato, A Heidari, A Mitra (2018) The Creation of C13H20BeLi2SeSi . The Proposal of a Bio–Inorganic Molecule, Using Ab Initio Methods for The Genesis of a Nano Membrane. Arc Org Inorg Chem Sci 3(4).
- A L Frischknecht, L J D Frink (2009) Bio membrane frontiers: nanostructures, models, and the design of life. Humana Press.
- Dennis Chapman (eds.), Biomembrane Structure and Function (Topics in Molecular and Structural Biology) Hardcover-1983.
- IN Levine (2003) Quantum Chemistry. Pearson Education (Singapore) Pte. Ltd., Indian Branch, India, 5th ed. 2003.
- A Szabo, NS Ostlund (1989) Modern Quantum Chemistry. Dover Publications, New York.
- P Hohenberg, W Kohn (1964) Inhomogeneous electron gas. Phys Rev 136(3B): B864–B871.
- W Kohn, LJ Sham (1965) Self-consistent equations including exchange and correlation effects. Phys. Rev 140(4A): A1133-A1138.
- JP Perdew, M Ernzerhof, K Burke (1996) Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys 105(22): 9982–9985.
- TH Dunning Jr (1989) Gaussian basis sets for use in correlated molecular calculations, The atoms boron through neon and hydrogen. J Chem Phys (90): 1007–23.
- RA Kendall, TH Dunning Jr, RJ Harrison (1992) Electron affinities of the first-row atoms revisited. Systematic basis sets and wave function. J Chem Phys (96): 6796–806.
- DE Woon, TH Dunning Jr (1993) Gaussian-basis sets for use in correlated molecular calculations. The atoms aluminum through argon. J Chem Phys (98): 1358–71.
- KA Peterson, DE Woon, TH Dunning Jr (1994) Benchmark calculations with correlated molecular wave functions. The classical barrier height of the H+H4 –¿ H4+H reaction. J Chem Phys 100(10): 7410–7415.
- AK Wilson, T van Mourik, TH Dunning Jr (1996) Gaussian basis sets for use in Correlated Molecular Calculations. Sextuple zeta correlation consistent basis sets for boron through neon. J Mol Struct (Theochem) 388(11): 339–349.
- MS Gordon, Kim K. Baldridge, Jerry A. Boatz, Steven T. Elbert, Mark S. Gordon et al., (1993) General atomic and molecular electronic structure system (GAMESS). J Comput Chem 14(11): 1347–1363.
- MS Gordon, MW Schmidt (2005) Advances in electronic structure theory: GAMESS a decade later. Theory and Applications of Computational Chemistry: the first forty years” Elsevier. CE Dykstra, GF renking, KS Kim, GE Scuseria (Edts.), Amsterdam, pp: 1167–1189.
- A Schaefer, H Horn, R Ahlrichs (1992) Fully optimized contracted Gaussian-basis sets for atoms Li to Kr. J Chem Phys 97: 2571-2577.
- A Schaefer, C Huber, R Ahlrichs (1994) Fully optimized contracted Gaussian-basis sets of triple zeta valence quality for atoms Li to Kr. J Chem. Phys 100: 5829-5835.
- F Weigend, R Ahlrichs (2005) Balanced basis sets of split valences, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys 7(18): 3297-3305.
- R Gobato (2017) Study of the molecular geometry of Caramboxin toxin found in star flower (Averrhoa carambola L.). Parana J Sci Edu 3(1): 1-9.
- R Gobato, A Gobato, DFG Fedrigo (2015) Molecular electrostatic potential of the main monoterpenoids compounds found in oil Lemon Tahiti - (Citrus Latifolia Var Tahiti). Parana J Sci Edu 1(1): 1-10.
- R Gobato, DFG Fedrigo, A Gobato (2015) Allocryptopine, Berberine, Chelerythrine, Copsitine, Dihydrosanguinarine, Protopine and Sanguinarine. Molecular geometry of the main alkaloids found in the seeds of Argemone Mexicana Linn. Parana J Sci Edu 1(2): 7-16.
- R Gobato, A Heidari (2018) Infrared Spectrum and Sites of Action of Sanguinarine by Molecular Mechanics and ab initio Methods. International Journal of Atmospheric and Oceanic Sciences 2(1): 1-9.
- R Gobato, DFG Fedrigo, A Gobato (2015) Molecular geometry of alkaloids present in seeds of mexican prickly poppy. Cornell University Library. Quantitative Biology.
- R Gobato, A Gobato, DFG Fedrigo (2016) Study of the molecular electrostatic potential of D-Pinitol an active hypoglycemic principle found in Spring flower Three Marys (Bougainvillea species) in the Mm+ method. Parana J Sci Educ 2(4): 1-9.
- R Gobato, A Heidari (2017) Calculations Using Quantum Chemistry for Inorganic Molecule Simulation BeLi2SeSi. Science Journal of Analytical Chemistry 5(5): 76-85.
- R Gobato, A Heidari (2017) Calculations Using Quantum Chemistry for Inorganic Molecule Simulation BeLi2SeSi. Science Journal of Analytical Chemistry 5(6): 76-85.
- R Gobato, A Heidari (2018) Molecular Mechanics and Quantum Chemical Study on Sites of Action of Sanguinarine Using Vibrational Spectroscopy Based on Molecular Mechanics and Quantum Chemical Calculations. Malaysian Journal of Chemistry 20(1): 1-23.
- A Heidari, R Gobato (2018) A Novel Approach to Reduce Toxicities and to Improve Bioavailabilities of DNA/RNA of Human Cancer Cells–Containing Cocaine (Coke), Lysergide (Lysergic Acid Diethyl Amide or LSD), Δ⁹– Tetrahydrocannabinol (THC) [(–)–trans–Δ⁹–Tetrahydrocannabinol], Theobromine (Xantheose), Caffeine, Aspartame (APM) (NutraSweet) and Zidovudine (ZDV) [Azidothymidine (AZT)] as Anti–Cancer Nano Drugs by Coassembly of Dual Anti–Cancer Nano Drugs to Inhibit DNA/ RNA of Human Cancer Cells Drug Resistance. Parana Journal of Science and Education 4(6): 1-17.
- A Heidari, R Gobato (2018) Ultraviolet Photoelectron Spectroscopy (UPS) and Ultraviolet–Visible (UV–Vis) Spectroscopy Comparative Study on Malignant and Benign Human Cancer Cells and Tissues with the Passage of Time under Synchrotron Radiation. Parana Journal of Science and Education 4(6): 18-33.
- R Gobato, A Heidari (2018) Using the Quantum Chemistry for Genesis of a Nano Biomembrane with a Combination of the Elements Be, Li, Se, Si, C and H. J Nanomed Res 7 (4): 241-252.
- R Gobato (2017) Study of the molecular geometry of Caramboxin toxin found in star flower (Averrhoa carambola L.). Parana J Sci Educ 3(1): 1-9.
- R Gobato, A Gobato, DFG Fedrigo (2015) Molecular electrostatic potential of the main monoterpenoids compounds found in oil Lemon Tahiti – (Citrus Latifolia Var Tahiti). Parana J Sci Educ 1(1): 1-10.
- R Gobato, DFGFedrigo, A Gobato (2015) Allocryptopine, Berberine, Chelerythrine, Copsitine, Dihydrosanguinarine, Protopine and Sanguinarine. Molecular geometry of the main alkaloids found in the seeds of Argemone Mexicana Linn. Parana J Sci Educ 1(2): 7-10.
- R Gobato, A Heidari (2018) Infrared Spectrum and Sites of Action of Sanguinarine by Molecular Mechanics and ab initio Methods. International Journal of Atmospheric and Oceanic Sciences 2(1): 1-9.
- R Gobato, DFG Fedrigo, A Gobato (2015) Molecular geometry of alkaloids present in seeds of mexican prickly poppy”, Cornell University Library. Quantitative Biology.
- Gobato, A Gobato, DFG Fedrigo (2016) Study of the molecular electrostatic potential of D–Pinitol an active hypoglycemic principle found in Spring flower Three Marys (Bougainvillea species) in the Mm+ method. Parana J Sci Educ 2(4): 1-9.
- DFG Fedrigo, R Gobato, A Gobato (2015) Avrocar: a real flying saucer. Cornell University Library.
- Creative Commons, (CC BY 4.0), https://creativecommons.org/licenses/ by/4.0/. “List of Intel Core i3 microprocessors”, https://en.wikipedia. org/wiki/List_of_Intel_Core_i3_microprocessors, Available in: August 30, 2018.
- The Cambridge Crystallographic Data Centre (CCDC), “Mercury - crystal structure visualisation, exploration and analysis made easy”, May 2012. Mercury 3.1 Development (Build RC5). The Cambridge Crystallographic Data Centre.
- Avogadro: an open-source molecular builder and visualization tool. 2012, Version 1.1.1.
- D Marcus, DE Hanwell, DC Curtis, TV Lonie, E Zurek, et al. (2012) Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. Journal of Cheminformatics 4: (17).
- Creative Commons, (CC BY 4.0), https://creativecommons.org/licenses/ by/4.0/. “Ubuntu (operating system)”, https://en.wikipedia.org/wiki/ Ubuntu_(operating_system), Available in: Aug. 31, 2018.
- Origin Lab 2018 Evaluation Licence, Graphing & Analysis, ©Origin Lab Corporation, 2018.


 We use cookies to ensure you get the best experience on our website.
We use cookies to ensure you get the best experience on our website.