Mini Review 
![]() Creative Commons, CC-BY
Creative Commons, CC-BY
Physics-Informed AI Infers Ligand-Induced Folding in Target Proteins
*Corresponding author:L Ridgway Scott, University of Chicago, Chicago, Illinois 60637.
Received: March 14, 2024; Published: March 21, 2024
DOI: 10.34297/AJBSR.2024.21.002902
Abstract
We propose a hybrid, physics-informed, AI approach to enhance the impact of homology modeling (AlphaFold2) for the identification of targetable structures in drug-discovery. This approach remedies deficiencies in structural databases due to underrepresented regions associated with structural plasticity.
Keywords: Alphafold2, Drug design, Drug-target complex, Induced protein folding, Physics-informed AI, Structural plasticity
Conformational Plasticity in Drug Design
Predicting ligand-induced protein folding, the structural adaptation of a target to a drug ligand, is one of the greatest challenges today in drug design [1]. The field has undergone a revolution due to the introduction of AlphaFold [2,3], the most accurate predictor of target structure from sequence. Like all machine-learning tools, AlphaFold is limited by the available structural databases. The protein structures in drug-target associations involving new compounds and targets are significantly under-represented in structural databases. This is because ligand associations often involve some structural deficiencies in a target protein.
This situation can be mitigated with physics-informed AI [4], whose purpose is to compensate for regions of data space that are poorly represented. Structural plasticity corresponds to regions of conformational diversity. Such regions are under-represented in repositories of structural data. A physical model that accounts for such plasticity is the dehydron [5], a backbone hydrogen bond unshielded from solvent. Dehydrons are naturally under-represented in structural data bases since they lie in the tails of stability distributions of hydrogen bonds. Using a combination of structural biology and physical chemistry concepts, dehydrons have been identi ed in protein structures [5-7]. The biophysics concept of dehydron provides a way to compensate for the lack of diversity in existing protein-structure data bases, as we describe here.
Plasticity does not mean disorder. However, software to predict disorder, such as PONDR [8], can be used to predict plasticity, by considering regions where there is a transition from order to disorder. Such regions are rich in dehydrons [9], where structural plasticity is likely, and have been dubbed the so-called structural twilight zones [9,10]. Due to their solvent exposure, dehydrons support plasticity because they are less strong and stable than well shielded backbone hydrogen bonds.
Structural plasticity provides significant targets for drug designers. For example, most signaling proteins have regions of plasticity, storing conformational entropy that makes their interactions ephemeral and weak, and hence difficult to target. Moreover, almost two-thirds of enzymes show conformational changes on binding their ligands [1].
In a previous paper [11], a proposal was made to augment AlphaFold’s internal data representation to allow for the incorporation of dehydron patterns. Here we suggest how PONDR and Al phaFold can be combined in a pipeline to indicate where structural plasticity may exist. These limited regions of plasticity can then be optimized to suggest different structures that possess significant affinity for drug leads. PONDR and AlphaFold have been used together before, and they predict similar disordered regions [12], but PONDR has not been used before to indicate plasticity.
Alphafold2 Misses the Targetable Structure
(Figure 1) The following example [1] illustrates the need for integrating plasticity signals into the structural inference of protein- ligand complexes. AlphaFold2 predicts correctly the unbound structure of the multi-domain protein Pro X from Archaeon globus fulgidus at RMSD 3.8 (PDB ID 1SW5). But the bound state (PDB ID 1SW1) differs substantially from the prediction. Figure 1 depicts a region of high plasticity that enables the binding event. Not surprisingly, there are dehydrons (marked in green) buttressing the bound-state (Figure 1(b)), a conformation not seen in the top AlphaFold2 predictions (Figure 1(a)). The predicted free conformation in the plastic zone is more rigid as evidenced by a higher number of water-shielded backbone hydrogen bonds, shown as grey lines. This is expected by the under-representation of dehydrons in structural databases.

Figure 1: The plastic parts of the multi-domain protein ProX from Archaeoglobus fulgidus. (a): the apo state, PDB ID 1SW5 (b): the bound state PDB ID 1SW1. A simplified α-carbon representation of the backbone (blue tubes) is presented. Amide-carbonyl hydrogen bonds are indicated as lines joining the paired residues, grey for well-shielded hydrogen bonds and green dehydrons.
A signal for plasticity can be inferred from disorder scores of the sequence. In Figure 2 we give the PONDR disorder score for the sequence of PDB ID 1SW5/1. Marked by a box in Figure 2 is the region A205-Glu to A213-Pro of plasticity determined by a sharp transition between order and disorder. These twilight regions are limited in extent, and this allows an approach to structural optimization of drug targets using simpler, exhaustive conformation-sampling strategies.

Figure 2: The PONDR score (13 July 23 download from http:www.pondr.com) [8] with the plasticity region A205-A213 highlighted.
Filling The Gaps in Structural Databases
Thus we propose a hybrid, physics-informed, AI approach to structure prediction. We use homology modeling (AlphaFold2) to predict a base structure. We then find the twilight zones of possible plasticity in this base structure, e.g., by using PONDR. The resulting small number of plastic deformations of the base structure can then be examined exhaustively to look for structures of interest, that can provide complexes of pharmacological impact. Our hybrid scheme uses an enhanced view of protein structure that splits proteins into three categories:
a) elastic (structured),
b) plastic, and
c) disordered (unstructured),
where the plastic zones are identified as twilight zones, using the dehydron concept. For proteins whose plastic zones are small, their possible structures are easily examined for relevant structural modifications. This translation of a physical concept into a computational algorithm will broaden the technological base for drug discovery.
Local Optimization
Since the plasticity regions are small, simple optimization schemes can be used to exhaustively search out all possibilities. One approach uses the Ramachandran Basins [13] to predict folded structure. These authors were able to fold correctly the villin headpiece, a 36-residue protein. Thus a much smaller region, such as a plastic region, can be done in reasonable time.
A major determinant of protein structure is the Backbone Hydrogen Bond (BHB) network. Although unstructured regions will often not have any BHBs, we assume that a plastic region has BHBs. We can describe the BHB network by a matrix M, indexed by residue number, with non-zero values where there is a BHB linking two residues. For the two structures 1SW1 and 1SW5, the corresponding BHB matrices for the plastic region indicated in Figure 2 are given in equations (1) and (2):

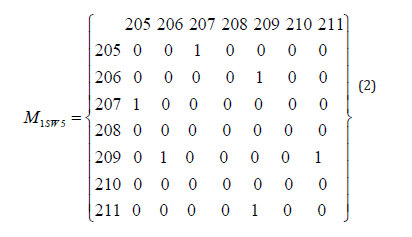
These matrices can be viewed as constraints on the protein structure. In carrying out structural optimization, such constraints could be used to limit the search space. Further constraints include potential clashes for the remainder of the structure that remains unchanged, due to rotations that occur at the ends of the plastic region.
The search space can be further reduced by reducing the number of BHB matrices considered. Typical constraints on the BHB matrices M include that Mij = 0 for |i-j| ≤1. By definition, they are symmetric. Thus for the matrices given in equations (1) and (2), the set of nonzero possibilities corresponds to a lower-triangular 5x5 matrix. The number of such matrices with only one nonzero is 15. Simple combinatorics yields the number of matrices with n nonzero as a function of n. Although this grows very fast with n, for small n it is a manageable set of possible BHB matrices.
If we are using Ramachandran basins in the optimization, the given BHBs give restrictions on the Ramachandran basins for the residues with nonzero BHB matrix entries. Putting together all of these constraints yields a manageable problem to solve computationally.
Data and Software Availability
The analysis in this paper was done on PDB files 1SW5 and 1SW1, both available at the Protein Data Bank, https://www.rcsb. org. Figure 1 was produced by Yapview, available at
https://sourceforge.net/projects/protlib/files/yapview/.
Figure 2 was produced by PONDR, available at http://www.pondr. com.
References
- Neera Borkakoti, Janet M Thornton (2023) AlphaFold2 protein structure prediction: Implications for drug discovery. Current opinion in structural biology 78: 102526.
- Minkyung Baek, David Baker (2022) Deep learning and protein structure modeling. Nature Methods 19(1):13-14.
- Ewen Callaway (2022) What's next for the AI protein-folding revolution. Nature 604: 234-238,
- Zhongkai Hao, Songming Liu, Yichi Zhang, Chengyang Ying, Yao Feng, et al. (2022) Physics-informed machine learning: A survey on problems, methods and applications. arXiv preprint arXiv: 2211.08064.
- Ridgway Scott, Ariel Fern ndez (2017) A Mathematical Approach to Protein Biophysics. Springer-Verlag.
- Carla Mattos, Cornelia R Bellamacina, Ezra Peisach, Antonio Pereira, Dennis Vitkup, et al. (2006) Multiple solvent crystal structures: probing binding sites, plasticity and hydration. Journal of Molecular Biology 357(5): 1471-1482.
- Eric J Sundberg, Roy A (2000) Mariuzza Luxury accommodations: the expanding role of structural plasticity in protein protein interactions. Structure 8(7): R137-R142.
- Zoran Obradovic, Kang Peng, Slobodan Vucetic, Predrag Radivojac, Celeste J Brown, et al. (2003) Predicting intrinsic disorder from amino acid sequence. Proteins: Structure, Function, and Bioinformatics 53(S6): 566-572.
- Ariel Fernandez (2016) Dehydron-rich proteins in the order-disorder twilight zone. In Physics at the Biomolecular Interface: Fundamentals for Molecular Targeted Therapy. Springer International Publishing pp.121-150.
- Natalia Pietrosemoli, Alejandro Crespo, Ariel Fernandez (2007) Dehydration propensity of order- disorder intermediate regions in soluble proteins. Journal of proteome research 6(9): 3519-3526.
- Ariel Fernandez (2020) Artificial intelligence teaches drugs to target proteins by tackling the induced folding problem. Molecular Pharmaceutics 17(8): 2761-2767.
- Gundeep Kaur, Ren Ren, Michal Hammel, John R Horton, Jie Yang, et al. (2023) Allosteric autoregulation of DNA binding via a DNA-mimicking protein domain: a biophysical study of ZNF410 DNA interaction using small angle X-ray scattering. Nucleic Acids Research 51(4): 1674-1686.
- Fernandez, Min yi Shen, Andr s Colubri, Tobin R Sosnick, R Stephen Berry, et al. (2003) Large-scale context in protein folding: villin headpiece. Biochemistry 42(3): 664-671.


 We use cookies to ensure you get the best experience on our website.
We use cookies to ensure you get the best experience on our website.